

 在线客服
在线客服
 快速发布
快速发布
 我的店铺
我的店铺
 我的化学加
我的化学加
 我的消息
我的消息
 我要充值
我要充值


 回到顶部
回到顶部



买产品

克难题

技术供需

求职招聘

发定制

项目整合

园区推荐

行业资讯

化学加智库

商家
买产品
克难题
技术供需
求职招聘
发定制
项目整合
园区推荐
行业资讯
化学加智库
商家
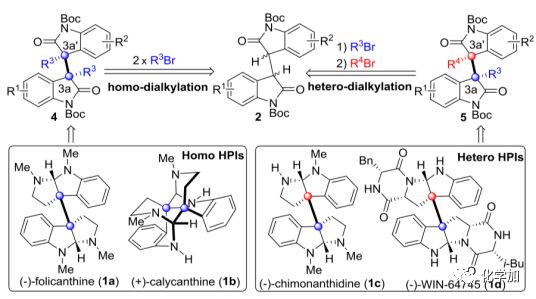
图1代表性的HPI天然产物以及本文的合成设计
(图片来源J. Am. Chem. Soc.)
如图1所示,许多天然二聚和低聚的六氢吡咯吲哚(HPI)生物碱(包括同型和异型)都具有重要的生物活性,如抗真菌和抑制细胞生长作用等。从结构上讲,这些分子通常在C3a和C3a’处包含一对具有空间位阻的季碳中心,从有机合成的角度来看,这是一个“艰巨的挑战”。事实上,对于这种结构基元的催化不对称合成非常困难,到目前为止仅有双Michael加成和双脱羧烯丙基化等方法,而且,这些报道的方法在收率和选择性方面并不高,它们只适用于同型生物碱的合成。因此,非常有必要开发一种更高效和通用的催化不对称二烷基化方法来合成这类结构基元。
在过去的几十年中,四级铵、三唑盐和相关的有机阳离子催化剂在2-氧化吲哚的单烷基化反应中起着重要作用。然而,对于二聚氧化吲哚同种和不同种二烷基化反应方面却报道甚少,一是可能在第二个烷基化过程中C3a和C3a’之间的空间位阻作用增加,可能导致意想不到的副反应,如非对映异构的烷基化和C3a-C3a’键断裂。另一个挑战在于如何获得不同类型的二烷基化反应(图1),这需要两种不同的亲电试剂(R3Br和R4Br)的反应性和立体可控性要能足够的匹配。因此,为了应对这些挑战,基于前期的工作基础,涂永强院士团队发展了一种新型的SPA(螺环吡咯酰胺)-三唑阳离子催化剂,成功地实现了二聚氧化吲哚的不对称同种和不同种二烷基化反应。
作者首先设计和制备了几种新型的SPA-三唑溴化铵催化剂(Cat 1-6,图2),并通过Cat 4的单晶证实了它们的绝对构象。以二聚氧化吲哚2a和4当量商业可得的溴乙酸苄酯3a为模型底物,研究了有机催化的不对称二烷基化反应。通过对各种溶剂、碱和添加剂的筛选,最终发现在3 mol% Cat 1(entry 1,74%收率,93% ee,12.6:1 dr)的条件下,甲苯中室温反应,就能较好地得到预期的同种二烷基化产物4aa。虽然其它催化剂(Cat 2-6)也能催化这一反应(entry 2-6),但结果较差。值得注意的是,N-甲基取代的Cat 6对映选择性最差(-6% ee,entry 6),表明在该不对称反应中,SPA型催化剂中N-质子的存在是立体选择性的必要条件。
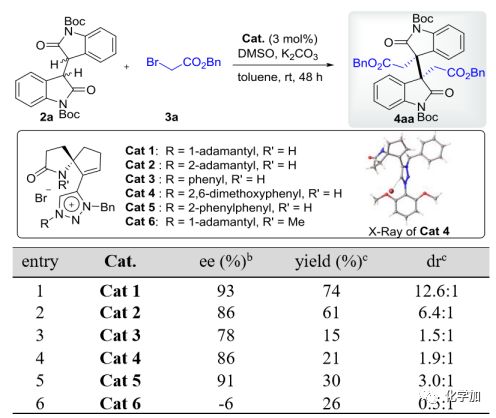
图 2 同种二烷基化反应条件的筛选
(图片来源 J. Am. Chem. Soc.)
得到最佳条件以后,作者对亲电试剂R3Br和2a的同种二烷基化反应进行了底物扩展。图3列出的结果表明,三组亲电试剂Ⅰ-Ⅲ(Ⅰ组:溴乙酸苄酯3a-i,II组:溴乙酸烷基酯3j-l和Ⅲ组:烯丙基溴代物3m和3n,均为4倍当量)都成功地得到了预期的同种二烷基化产物4aa-4an,在大多数情况下(entry 1-14)都取得了满意的结果。在第一组中,所有的亲电试剂3a-i(entry 1-9)都能顺利地反应,得到高对映选择性(88-95% ee)和良好的产率(63-89%)。这些亲电试剂的苄基取代基上电子效应对反应结果也有一定的影响,其中4-F-苄基酯3g的结果最好(entry 7)。对于第二组(entry 10-12),烷烃的位阻对反应效率的影响是非常明显的。例如,小位阻的甲酯3j仅在2天内就能反应得到4aj,且具有良好的立体选择性(entry 10),并由单晶确定了其结构和立体化学,而体积较大的乙基酯3k则需要更长的反应时间10天(entry 11)。此外,当将体积较大的正丁酯3l应用于该体系时,无法得到所需的产物4al(entry 12)。第三组中的烯丙基亲电试剂3m和3n对此反应也是可行的,但结果却不相同,3m的反应具有较高的ee和产率(entry 13),而3n则具有较高的dr和产率(entry 14)。
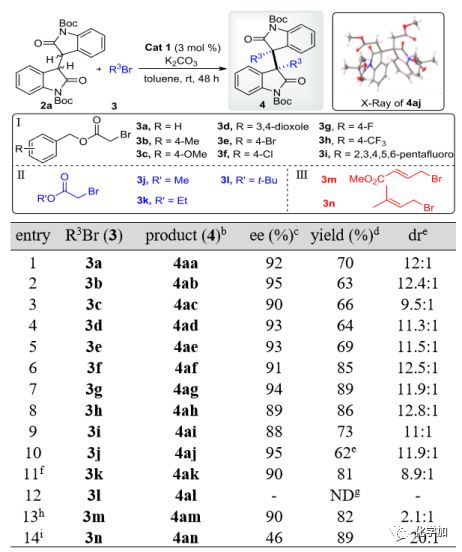
图 3 同种二烷基化中亲电试剂的考察
接着,作者以3a为亲电试剂,对多种芳环上单、双取代的二聚氧化吲哚类化合物2进行了考察。如图 4所示,该反应具有广泛的底物兼容性。对于C5a、C6a或C7a的单取代底物,吸电子取代基(2-5,7-9和12)比给电子取代基普遍有利,然而,C4a-Cl取代的底物不能得到所需产物,只能产生复杂的混合物,这可能是由于其对反应中心(C3a或C3a’)的空间屏蔽所致。在双取代底物(entry 13-18)中,非对称(entry 13-15)和对称(entry 16-18)双取代在此反应中都是有效的,且具有优良的对映选择性(92-98% ee)、良好到优良的非对映选择性(7.1:1至>20:1 dr)以及中等至良好的产率(46-85%)。与单取代底物类似,二取代物的电子性质对反应结果也有显著影响。例如,二氯代底物能快速反应生成相应的产物4ra,产率为85%,ee 值为92% (entry 17),而二甲氧基取代底物反应的结果相对较差(33%产率,98% ee,>20:1 dr),还需要添加DMSO作为极性共溶剂进一步改进,才能以较高的产率得到4sa(78%产率,92% ee,18:1 dr,entry 18)。重要的是,对于产物4ca和4da,可以通过C-C偶联反应和进一步转化来合成更复杂的三聚HPI生物碱,具有重要的应用价值。
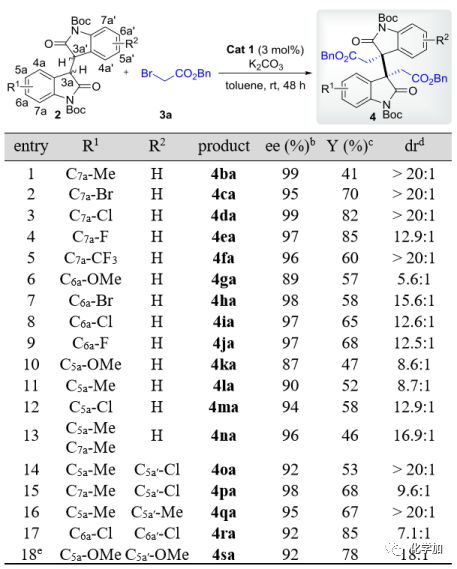
图 4 同种二烷基化反应中二聚氧化吲哚底物的扩展
完成了二聚氧化吲哚的同种二烷基化反应,接着作者用两种不同的亲电试剂R3Br和R4Br,研究了更具挑战性的一锅法不同种的二烷基化。最初作者发现在Cat 1(3 mol%)催化下, 1当量的3a和2a反应能得到单烷基化产物,化学选择性良好,产率约85%,没有检测到二烷基化产物。根据这一观察结果,在2a消失后,向反应体系中加入3当量的3g,就得到了所需的不同种二烷基化产物5a,产率54%,ee值96%。受此鼓舞,使用不同的亲电试剂R3Br, 作者又尝试了更多的不同种二烷基化反应,均能得到预期的产物5b-f。特别地,5c-f可作为关键中间体,用于合成复杂的HPI生物碱。

图 5 2a的不同种二烷基化反应研究
接着,作者对三唑催化的烷基化反应过程中的立体化学控制进行了探究,根据观察到的结果,作者提出了单烷基化和二烷基化反应的亲核进攻模型(图 6)。由于Cat 6没有酰胺N-H部分,仅给出了较差的立体选择性。而在Cat 1和烯醇式反应时,每一步烷基化过程中都可能存在氢键和离子对相互作用,由于金刚烷基大的位阻作用,中间体从背面向亲电试剂的亲核进攻是不利的,从而导致主要形成(S,S)-二烷基化产物。
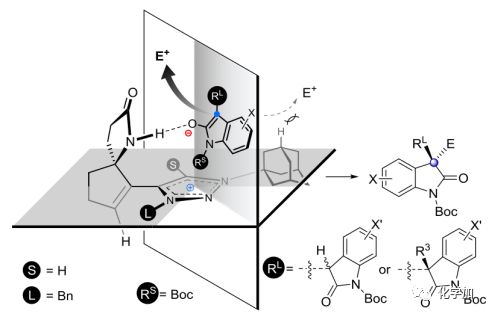
图 6 立体化学控制模型
为了验证该催化不对称二烷基化反应的有效性,作者还将其用于了天然产物的全合成中,实现了(–)-folicanthine (1a)的形式全合成和(–)-chimonanthidine (1c)的首次不对称全合成。在(1c)的合成中,主要挑战在于在两个吡咯烷环存在下,如何选择性地用甲基仅保护一个N-H。而用不同种二烷基化产物5c(可以克级规模制备,重结晶后ee大于99%)作为关键中间体,只需要一次柱层析纯化,经脱Boc、甲基保护、胺解及还原胺化就可以有效地得到关键的合成子7,具有81%的总收率,区域选择性还原实现了吲哚部分的胺化/环化反应,随后化合物7的C=C键氧化裂解,得到醛化合物8,通过还原胺化反应得到产物9,再一次还原胺化/环化反应生成第二个吡咯烷环,最后在Pd(OH)2/C催化条件下脱去苄基保护基,就实现了(–)-chimonanthidine (1c)的首次不对称全合成。
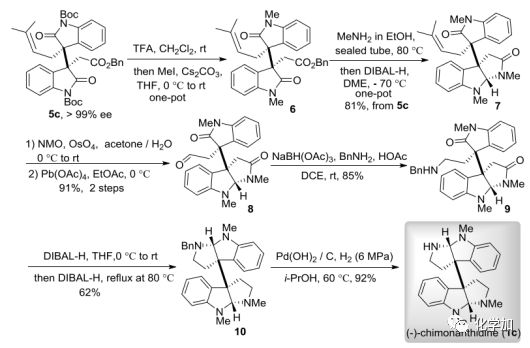
图 7(–)-chimonanthidine的不对称全合成
总结:涂永强院士团队成功地基于氮杂螺环骨架开发了SPA三唑阳离子催化剂,并利用该催化剂实现了二聚氧化吲哚类化合物的不对称同种和不同种二烷基化反应,该反应操作简便、条件温和,可以高效地构建极具挑战性的相邻季碳中心(收率高达82%,ee高达99%,具有>20:1 的dr)。到目前为止,该转化是构建这类合成基元的最有效的方法。此外,从产物5c出发,可以很容易地实现(–)-chimonanthidine (1c)的不对称全合成,体现了该方法的重要应用价值。
声明:化学加刊发或者转载此文只是出于传递、分享更多信息之目的,并不意味认同其观点或证实其描述。若有来源标注错误或侵犯了您的合法权益,请作者持权属证明与本网联系,我们将及时更正、删除,谢谢。 电话:18676881059,邮箱:gongjian@huaxuejia.cn
