过渡金属参与的不对称催化氢化反应是获得手性分子最高效的途径之一,而杂环化合物的不对称氢化一直以来都是该领域的研究热点。特别是对于苯并杂环类化合物,其很强的稳定性导致氢化反应条件较为苛刻,而刚性结构更是影响了底物与催化剂的有效配位,其高效不对称催化氢化更是成为该领域的挑战性课题。上海交通大学张万斌教授课题组长期从事过渡金属催化的不对称催化氢化反应研究,并取得了一些可喜的进展(近三年代表性工作:Nat. Chem. 2022, 14, 920. Nat. Sci. 2021, e10021. Angew. Chem. Int. Ed. 2021, 60, 23602; 2021, 60, 16989; 2020, 59, 5371; 2019, 58, 15767; 2019, 58, 11505; 2019, 58, 7329. Nat. Commun. 2020, 11, 5935)。2021年,该课题组首次利用BridgePhos–Rh催化体系实现了具有刚性结构内酯型香豆素的高效不对称催化氢化,以高达98%的收率和99.7%的对映选择性获得了手性3-氨基二氢香豆素。通过单晶结构发现在不改变其它因素的前提下,仅通过改变配体碳链的长度,可方便地调节BridgePhos–Rh配合物中联苯骨架的二面角,使配位膦原子上的轴向苯环与联苯骨架上的苯环具有最强的π-π相互作用,从而提供了最为合适的配位环境,并因此取得了优异的不对称氢化效果(Angew. Chem. Int. Ed. 2021, 60, 23602-23607)。最近,该课题组利用上述建立的BridgePhos–Rh催化体系,成功地实现了刚性3-氨基色酮衍生物的高效不对称连续催化氢化,机理研究表明该反应是经历了一条非常规的动态动力学拆分过程(图1)。图1 BridgePhos–Rh催化3-氨基色酮衍生物的不对称连续催化氢化作者以3-苯甲酰胺色酮(1a)作为模板底物进行反应条件优化,确定最优反应条件为:以[Rh(cod)2]SbF6/(S)-C10-BridgePhos为催化剂,二氯甲烷为溶剂,氢气压力40 atm,温度30 ℃,反应24 h。在最优反应条件下,一系列含有不同取代基的3-氨基色酮底物均以高收率和优异的立体选择性获得了氢化产物(图2)。为了进一步了解反应的过程,作者首先开展了反应动力学实验(图3)。发现在反应初期原料1a中的C=C双键被快速氢化,生成中间体2a,同时伴随着少量3a的生成。当反应进行到2 h时,原料1a只剩下16%,中间体2a产率达到最高值59%。随后2a被快速消耗,直至12 h反应完全。在反应最初的2 h内,中间体2a的ee值变化很小,随后逐渐降低,说明在该催化体系下(S)-2a中的碳氧双键还原速率快于(R)-2a。在整个反应过程中,3a的ee值没有明显的变化。如果将(S)-2a的ee值,通过反应时间的曲线外推到起始0点时,可以得出反应初始生成(S)-2a的ee值约为76%(图3a)。同样,将具有吸电子取代基的底物1h应用于上述的动力学实验,得到了与底物1a相似的结果。从整个动力学控制实验来看,化合物1h具有很高的反应活性,反应在90 min能够完全转化。同样通过反应时间的曲线外推到0点时,可以得出反应初始生成(S)-2h的ee值约为84%(图3b)。分析发现,化合物1a第一次氢化生成的2a起始ee值为76%,即(S)-2a与(R)-2a的相对含量分别为88%和12%,或者说反应起始时(S)-2a的含量为88%。根据反应结束后所有异构体的含量计算,生成3a的3-位为S-构型异构体的含量约为94.3%,即反应结束后该异构体的含量较反应起始(S)-2a的含量有了6.3%的提高(94.3% vs 88%)。这一结果说明反应过程中约有6.3%的(R)-2a转化为(S)-2a。在对1h用同样的方法进行分析时发现,大概有5.2%的(R)-2h在第二次氢化过程中转化为(S)-2h。以上结果均说明在1a或1h的不对称连续催化氢化过程中,第二次氢化可能存在动态动力学拆分过程(图3c)。

为了进一步阐明动态动力学拆分过程,作者进行了一系列其它控制实验。首先,在标准反应条件下,对消旋的中间体2a进行氢化。以上述同样的方式,对反应结束后的各个异构体含量进行分析,可以得出反应过程中有5%的(R)-2a转化为(S)-2a。而对消旋的2h进行氢化,发现有22%的(R)-2h转化为(S)-2h。但是在单一构型的2a和2h氢化过程中,均未观察到消旋化的现象。很明显,对第一次氢化反应的中间体2来说,只有当两个对映异构体同时存在时才能发生消旋化(图4)。

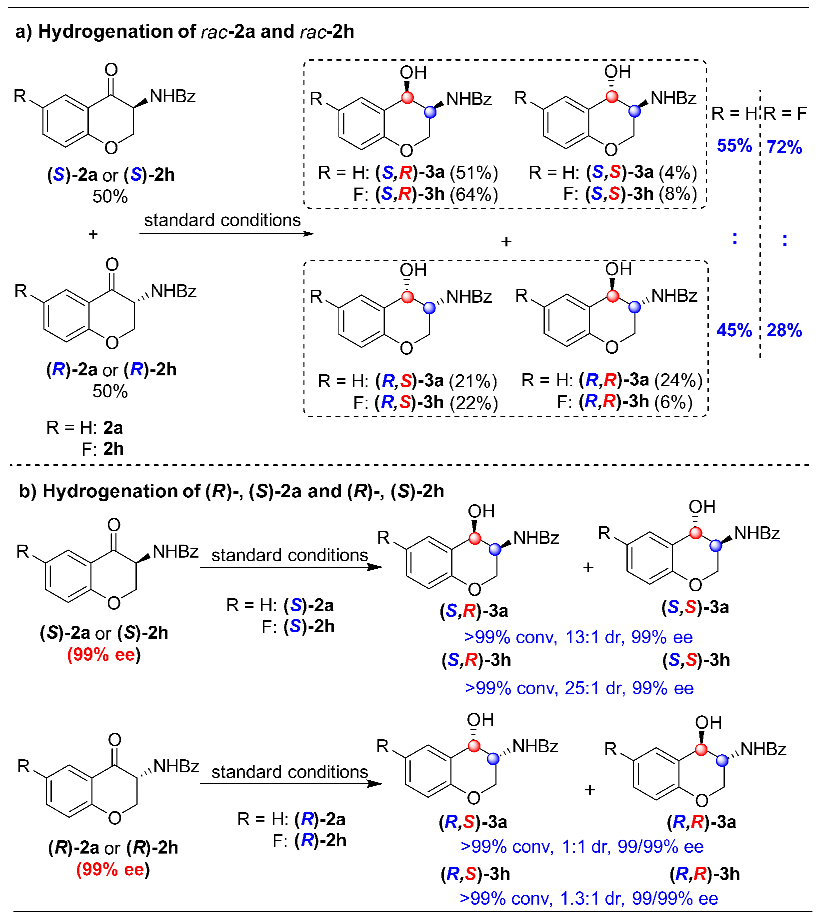
图4 控制实验:消旋和对映纯1a/1h的不对称催化氢化为了进一步搞清消旋化过程,作者开展了氘代实验研究。在标准反应条件下,将摩尔比为85:15的非氘代(S)-2h与氘代(R)-2a混合物应用于上述氢化反应。结果显示,除了得到氢化产物(S,R)-3h外,也观察到(S)-2h与氘代(R)-2a有明显的消旋化现象。需要指出的是,(R)-2a的3-位手性中心上的氘原子没有任何变化,而由其衍生而来的(S)-2a相应位置上的氘原子却完全被H原子替代,说明该消旋过程并非是以传统的烯醇互变形式进行的,很可能是以一种非常规的手性同化方式进行的(图5)。
图5 控制实验:非氘代(S)-2h与氘代(R)-2a混合物的不对称催化氢化通过在控制实验的基础上结合DFT计算,作者提出了反应的可能机理(图6)。第一步氢化的具体过程为:首先Rh催化剂经氢气活化,生成Rh(I)-氢气络合物,并与底物配位生成中间体0,然后其通过氢气分子氧化加成和氢原子迁移插入的协同过程生成中间体2,最后中间体2经还原消除得到中间体3。此后,中间体3可以与另一分子氢气结合并再次发生氧化加成生成中间体4。随后,4能够通过配位解离平衡释放出(S)-2a或者(R)-2a。DFT计算发现,(S)-2a可以继续发生H迁移插入,生成中间体5a,并接着与氢气结合,发生氢气异裂,氢质子转移到氧原子上生成中间体6a,最后解离出产物,完成第二次氢化过程。然而计算表明(R)-2a在进行第二次氢化时需要很高的活化能,极大地阻碍了其二次氢化反应的进行。因此(R)-2a更倾向于经过一个消旋化途径转换为(S)-2a从而完成二次氢化。其具体消旋化过程为:(R)-2a首先与中间体5a结合,生成六配位的铑中间体7a。由于中间体7a的醇阴离子具有较强的碱性,可以攫取(R)-2a立体中心上的氢原子,并使得(R)-2a的手性中心可以通过烯醇型中间体8a发生消旋化。考虑到导向基团的羰基与中心金属Rh的配位解离平衡,8a和8b可以通过中间体8c相互转换。最后,中间体8b中羟基的质子发生转移而形成7b,并解离生成中间体5a和(S)-2a,实现了(R)-2a到(S)-2a的单向转化。该过程表现为一个手性同化过程,而不是传统的消旋化过程。

接下来作者进行了该不对称连续氢化反应的放大实验,在底物/催化剂最高为500/1的摩尔比下,反应仍可高收率、高立体选择性地合成手性3-氨基-4-色烷醇,该产物可以被进一步衍生转化为多种药物活性中间体(图7)。

该氢化反应的中间体(S)-2a(可由3a氧化而得到)也是手性新药物开发过程中非常有用的合成砌块,通过一系列转化可得到多样化的手性天然产物或临床新药(图8)。

以上成果近期发表在国际著名期刊《美国化学会杂志》(Journal of the American Chemical Society, 2022, DOI:10.1021/jacs.2c09266)上,上海交通大学张万斌教授为该论文的通讯作者,上海交通大学药学院博士生徐运楠和上海交通大学化工学院博士生罗亦聪为共同一作,该项工作主要得到了国家自然科学基金,国家重点研发项目基金和上海市教委基金的资助。


 在线客服
在线客服
 快速发布
快速发布
 我的店铺
我的店铺
 我的化学加
我的化学加
 我的消息
我的消息
 我要充值
我要充值


 回到顶部
回到顶部















